

Üretken Yapay Zeka, AlphaFold ve RosettaFold gibi olağanüstü ilerlemelerle, ilaç keşif süreçlerini dönüştürmektedir. Biyoteknoloji şirketleri ve araştırma laboratuvarları protein yapılarını incelerken, protein etkileşimleri hakkında devrim niteliğinde bilgiler elde etmektedir.
Proteinler dinamik varlıklardır. Bir proteinin doğal durumu yalnızca amino asit dizilimi tarafından belirlenir; örneğin AlphaFold2 ve Uygulamaları üzerine yapılan araştırmalarda bu durum ortaya konmuştur. Ancak tek bir protein, çevresel koşullara bağlı olarak farklı yapısal konformasyonlar benimseyebilir. Bu konformasyonlar, esnek bölgeler, protein kompleksleri içindeki etkileşimler veya aktif ve inaktif durumlar arasındaki geçişleri yansıtabilir.
Protai, NVIDIA Inception programı kapsamında yer alarak, farklı protein durumları arasındaki yapısal değişiklikleri yakalamayı hedeflemektedir. Bu yaklaşım, belirli bir etki mekanizması için en doğru protein yapısını belirlemeye yönelik olup, en terapötik açıdan uygun olana yöneliktir.
Protai, İlaç keşfini ilerletmek için kütle spektrometrisi proteomik ve yapay zeka kullanarak topluma ve insan sağlığına önemli katkılarda bulunan hassas tıp çözümleri geliştirmektedir. Protai’nin platformunun merkezinde, Nobel ödüllü protein yapı algoritmalarını, fizik tabanlı araçları ve özel proteomik verileri birleştiren bir protein yapı tahmin hattı bulunmaktadır.

Protai, AI çıkarım sürelerini hızlandırmak amacıyla, NVIDIA NIM mikro hizmetlerini benimsemiştir. Bu optimizasyonlar, protein yapı tahminlerinde önemli ölçüde daha yüksek bir throughput ve daha düşük bir gecikme süresi sağlamıştır. Bu gönderide, Protai’nin, ilaç keşif süreçlerini dönüştüren doğru ve ölçeklenebilir protein yapı tahminlerini nasıl güçlendirdiğine dair detaylara yer verilecektir.
Protein Kompleks Yapı Tahmini Arka Planı
Protein komplekslerini anlamak, yapısal biyolojinin temel unsurlarından birisidir. Bir protein kompleksi, belirli biyolojik aktiviteleri gerçekleştirmek üzere etkileşimde bulunan iki veya daha fazla polipeptid zincirinin grubudur. Çoklu proteinlerden oluşan multimerik yapılar, kritik biyolojik süreçleri yönlendirmekte ve ilaç keşfindeki temel hedefler olarak öne çıkmaktadır. Bilim dünyası her bir protein monomerinin yapısını belirleme konusunda ilerleme kaydediyor olsa da, protein komplekslerinin sayısı katlanarak artmaktadır. Bu durum, yapısal çalışmalarda tahmin algoritmalarının önemini ortaya koymaktadır.
AlphaFold-Multimer, multimerik protein yapılarına yönelik yüksek kaliteli hesaplamalı tahminler yapmamıza olanak tanıyan bir yeniliği temsil ediyor. AlphaFold’un temelleri üzerine inşa edilen bu yenilik, derin öğrenmeyi kullanarak proteinler arasındaki etkileşimleri çözümler. AlphaFold algoritması, parametreleri, çıktıları ve dağıtımı hakkında daha fazla detay ise ilerleyen kısımlarda ele alınacaktır.
Protai, çok yönlü bir yaklaşım benimsemektedir. Ana stratejilerden biri, AlphaFold-Multimer gibi yapısal tahmin modellerinin örneklemesini artırarak yapısal kaymaları hesaba katmaktır. Ayrıca Protai, belirli protein yapılarını farklı durumlarda ortaya koyan, benzersiz çapraz bağlama kütle spektrometrisi (XL-MS) verileri üretmektedir.
XL-MS, kimyasal çapraz bağlayıcılar kullanarak belirli amino asit kalıntılarını proteinler içinde veya arasında kovalent olarak bağlayarak, mekânsal yakınlık ve etkileşim alanlarını elde eden güçlü bir deneysel tekniktir. Bu çapraz bağlı bölgeler değerli mesafe kısıtlamalarını sağlayarak protein konformasyonlarını ve etkileşimlerini daha hassas bir şekilde haritalamayı mümkün kılar. Deneysel olarak elde edilen kısıtlamaları gelişmiş örnekleme teknikleri ve moleküler dinamik simülasyonları ile birleştirerek, araştırmacılar kamuya açık verilerin ötesine geçebilen protein yapıları üretebilmektedirler.

Vaka Çalışması: H3-H4 Protein Kompleksinin Tahmini
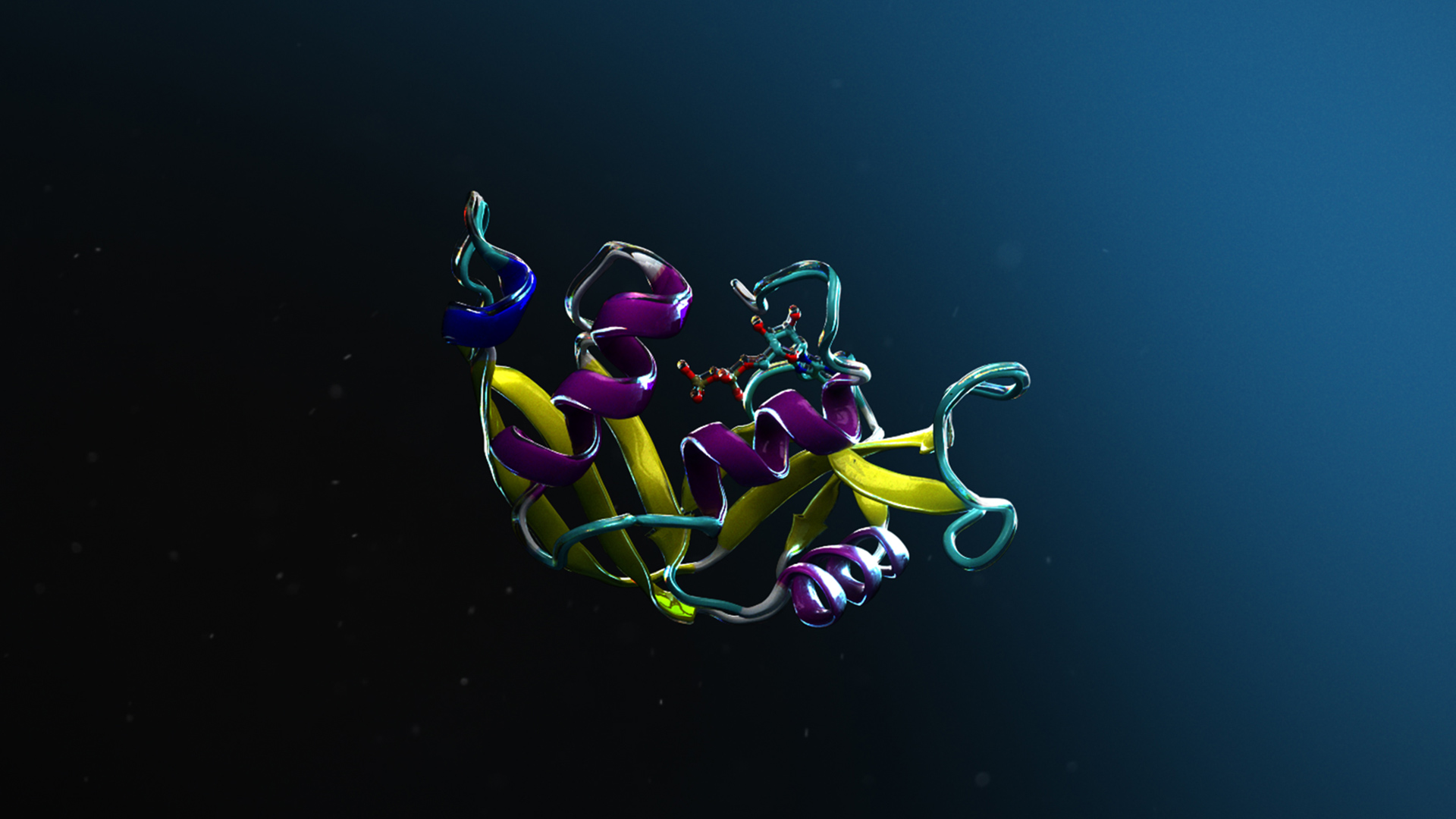
Protai’nin yeteneklerini sergilemek amacıyla bu bölüm, histon H3 ve H4’ün yer aldığı bir protein kompleksinin tahminini inceleyecektir. Bu kompleks, DNA’nın transkripsiyon faktörlerine ve RNA polimerazına erişilebilirliğini belirlemede önemli bir rol oynamaktadır ve ayrıca DNA’nın onarım süreçlerinde stabilitesine katkıda bulunmaktadır.
Protai, AlphaFold2-Multimer NIM kullanarak H3-H4 kompleksinin yapısal tahminini gerçekleştirmiştir. Elde edilen yapı, güven düzeyini gösteren renkli bir görselleştirme sunmaktadır. AlphaFold eğitim setinde yer alan proteinler için tahminler genellikle yüksek güvenle yapılmakta; ancak esneklik ve benzersiz özellikler belirli konformasyona bağlı olarak değişiklik göstermektedir.

Tahminleri hassaslaştırmak amacıyla Protai, H3 ve H4 arasındaki üç çapraz protein bağlayıcı linkeri belirlemiştir. Bu linkler, Protai’nin tahmin edilen yapıyı doğrulamasına veya yeni potansiyel protein durumlarını ortaya çıkarmasına olanak tanıyan deneysel bağlanma noktaları işlevi görmektedir. Bu durumda, en yüksek beş sıralı tahmin arasında üç link sürekli olarak korunmuştur. Ayrıca, iki proteindeki yüksek ve düşük güvenlikli kalıntılar arasındaki bölgede bir link tanımlanmış, bu da tahmini daha da geliştirmek için bir fırsat sunmuştur.

NVIDIA NIM ile AlphaFold-Multimer Dağıtımı
Protai, AlphaFold-Multimer ve diğer LLM tabanlı araçların ölçeklenebilir ve optimize edilmiş dağıtımını desteklemek için NVIDIA NIM kullanmaktadır. NIM, NVIDIA GPU’ları üzerinde kesintisiz dağıtım için optimize edilmiş çıkarım konteynerleri sağlar. Bu konteynerler, on-premises veya bulutta çalıştırıldığında en güncel performansı sunar. AlphaFold, tek bir tahmin için birden fazla GPU kullanamayabilir. Ancak NIM mikro hizmeti, birden fazla çıkarım görevinin paralel olarak çalıştırılmasını sağlar; bu da birçok protein kompleksinin tahmini için gereken süreyi önemli ölçüde azaltır (büyük kompleksler için tahmin süresi 24 saatten fazla olabilir).
Çalışma akışı şu ana aşamaları içermektedir:
- Çoklu Dizilim Hizalaması (MSA): MSA, etkileşimde bulunan proteinler arasındaki korunan bölgeleri ve birbirleriyle evrimsel sinyalleri belirler. Bu, tahminler için bir temel sağlar. Geleneksel MSA araçları, dizilim işlemlerini ardışık bir şekilde gerçekleştirdiklerinden, GPU’ların paralel işlem kapasitesine erişemez. AlphaFold2 NIM, MMseqs2 kullanarak, verimli karşılaştırmalara olanak tanıyan bir GPU-optimizasyonlu dizilim ve kümelenme araç setidir.
- Protein Etkileşimlerinin Modellenmesi: AlphaFold-Multimer, protein kompleks yapısı tahminine yönelik, etkileşimler açısından iyileştirilmiş temsilciler ve multimerik kayıplar dahil olmak üzere AlphaFold2 mimarisinin modifiye edilmiş bir versiyonunu kullanır.
- Yapı İyileştirme: Tahmin edilen yapılar, doğruluğun ve fiziksel geçerliliğin sağlanması amacıyla iyileştirilmektedir; bu süreçte stereokimyasal kısıtlamalar ve deneysel standartlar kullanılmaktadır.

AlphaFold2-Multimer NIM, çıkarım sürecini CPU ve GPU yoğun görevler arasında bölmek için birden fazla son nokta sunar; bu da hesaplama maliyetlerini azaltır:
- protein-structure/alphafold2/multimer/predict-structure-from-sequences: Diziden tam yapı tahmini (uçtan uca)
- protein-structure/alphafold2/multimer/predict-MSA-from-sequences: Dizi üzerinden MSA hesaplama (CPU yoğun)
- protein-structure/alphafold2/multimer/predict-structure-from-MSA: Önceden hesaplanmış bir MSA’den yapı tahmini (GPU yoğun)
Donanım seçimi, protein kompleksinin büyüklüğüne bağlıdır:
- Kısa diziler: 32 GB’lik bir GPU yeterli olacaktır.
- Daha büyük kompleksler (>3,000 kalıntı): Optimal performans için NVIDIA H100 veya A100 GPU’ları gereklidir.
Yukarıdaki kullanım örneği için Protai, H3 ve H4’ün küçük proteinler olması sebebiyle AlphaFold2-Multimer NIM’i bir NVIDIA L4 GPU’da özelleştirerek dağıtmıştır.
Parametreler
AlphaFold2-Multimer NIM, aşağıdaki parametrelere sahiptir:
- diziler: Çoklu yapısal tahminin yapılacağı hedef protein zincirlerini tanımlar.
- algoritma: Homolog dizilerin aranmasında kullanılan algoritmayı belirtir. jackhmmer, MSA oluşturma sürecinde yaygın olarak kullanılan bir algoritmadır; evrimsel olarak ilgili dizileri tanımlar ve modelin protein etkileşimlerini öğrenmesine yardımcı olur.
- e_değeri: Veritabanlarındaki homolog dizilerin belirlenmesi için bir eşik değeridir. Daha düşük değerler daha katı eşleşme kriterlerini ifade eder.
- iterasyonlar: MSA algoritmasının gerçekleştireceği iterasyon sayısını tanımlar (çalışma süresini sınırlar).
- veritabanları: Doğru MSA için gerekli evrimsel bilgilerin sorgulandığı veritabanlarını belirtir.
- rahatlama_tahmini: Tahmin edilen yapının rafine edilip edilmeyeceğini belirtir. Son yapının fiziksel geçerliliğini sağlamak amacıyla bağ uzunlukları, açıları ve stereokimyasal kısıtlamalar optimize edilir.
Çıktı
AlphaFold2-Multimer NIM çıktısı, Protein Veri Bankası (PDB) formatında tahmin edilen protein multimollerine dair atom düzeyindeki yapısal bilgileri içermektedir. Çıktı dosyasında, her bir atom, PDB formatı spesifikasyonuna uygun bir yapılandırılmış formatla tanımlanmıştır.

PDB çizgeleri, AlphaFold2-Multimer NIM ile aşağıdaki gibi açıklanmaktadır:
- Zincir tanımlayıcı: Çıktıda protein zincirini tanımlamak için kullanılır; örneğin, iki proteinden oluşan bir kompleks en az iki zincir içerir.
- Koordinatlar (X, Y, Z): Atomun 3D mekânsal koordinatları (Å cinsinden).
- Mevcudiyet: Bu atomun bu pozisyonda gözlemlenme olasılığıdır. 0.00 (tamamen yok) ile 1.00 (her zaman mevcut) arasında değişir. Deneysel verilerde, bir atomun bir kristal yapısında birden fazla konumda bulunması durumunda, mevcilliği 1.00 ‘dan az olabilir. AlphaFold tahminlerinde, mevcillik, proteinin en muhtemel yapısını sağlamak için tüm atomlar için 1 olarak ayarlanmıştır.
- B-faktörü: Yapının atomik yer değiştirmesi veya esnekliğini ölçen bir değerdir. Yüksek değerler daha fazla hareket anlamına gelir. Deneysel verilerde, bu durum sıcaklık hareketi veya düzensizliği temsil edebilir. AlphaFold çıktılarında, bakış açısı, pLDDT’den elde edilen bir güven puanı ile değiştirilmiştir.
Sonuç
Protai, AlphaFold2-Multimer NIM’i, deneysel olarak belirlenen benzersiz XL-MS linkleri ile birleştirerek yapı tahmin sürecini iyileştirmiştir. NVIDIA optimizasyonlu yapay zeka altyapısının avantajlarını kullanarak tahminler hızlandırılmış ve ölçeklendirilebilir hale getirilmiştir. Bu sayede daha önce erişilemeyen protein etkileşimleri keşfedilerek yeni ilaç keşfi ve hassas tıp alanında kapılar açılmıştır.
H3-H4 örneği, Protai’nin entegre yaklaşımının, yapısal tahmin ve XL-MS verilerini birleştirerek erişim sağladığı içgörüleri göstermektedir. Bu içgörüler, biyolojik işlevlerin ve terapötik uygulamaların anlaşılması için kritik öneme sahiptir.
Üretken yapay zekanın sürekli evrimiyle birlikte, NVIDIA NIM gibi araçlar, hesaplamalı biyolojinin tam potansiyelini açığa çıkarmada önemli bir rol oynayacaktır. Protai, en son yapay zeka teknolojilerini entegre etme taahhüdünü sürdürerek, hayat kurtarıcı ilaçların daha hızlı, daha verimli ve daha hassas bir şekilde geliştirilmesi için çalışmaya devam etmektedir.
İlaç keşfi için tüm NIM mikro hizmetleri setini deneyimleyebilir, NVIDIA BioNeMo İskeleti ile kendi biyoloji modellerinizi eğitebilir ve üretken sanal tarama için kullanabilirsiniz. Ayrıca, GPU hızlandırılmış MMseqs2 ile AlphaFold2 protein yapı tahminini nasıl artıracağınızı öğrenebilirsiniz.



