

Geçmişte, malzeme tasarımı son derece zahmetli bir işti. Bin yılı aşkın bir süre boyunca, araştırmacılar altın yapmak için kurşun, cıva ve kükürt gibi maddeleri bir araya getirmeye çalıştılar. Bu çaba, ünlü bilim insanları Tycho Brahe, Robert Boyle ve Isaac Newton gibi isimleri de kapsayan, sonuçsuz bir girişim olarak bilinen simyayı doğurdu.
Günümüzde, malzeme bilimi büyük ilerlemeler kaydetti. Son 150 yıldır, farklı elementlerin özelliklerini gösteren periyodik tablo‘nun sağladığı olanaklarla, elementlerin birinin diğerine dönüşmesinin mümkün olmadığını biliyoruz. Ayrıca, son on yılda makina öğrenimi araçları, çeşitli moleküllerin ve maddelerin yapısını ve fiziksel özelliklerini belirleme kapasitemizi önemli ölçüde artırdı. MIT’deki Malzeme Bilimi ve Mühendisliği profesörü Ju Li liderliğindeki bir grup tarafından yapılan yeni araştırma, malzeme tasarımını kolaylaştıracak önemli bir sıçrama vaadinde bulunuyor. Araştırma sonuçları, Aralık 2024 tarihli Nature Computational Science dergisinde yayımlanacak.
Yeni Hesaplama Yöntemleri
Bugün, moleküler sistemlerin karakterizasyonunda kullanılan çoğu makine öğrenme modeli, moleküllerin veya kristallerin toplam enerjisini elektron yoğunluğu dağılımına bakarak belirleyen yoğunluk fonksiyonel teorisi (DFT)‘ne dayanmaktadır. Ancak bu yaklaşımın bazı dezavantajları vardır. Li’ye göre, “Öncelikle, doğruluğu her zaman mükemmel değil. İkincisi ise, size sadece bir şey söylüyor: moleküler sistemin en düşük toplam enerjisi.”
“Çift Terapi” ile Çözüm
Li’nin ekibi, bu problemleri aşmak için kuplaj kümesi teorisi olarak bilinen farklı bir hesaplama kimyası tekniğine yöneldi. “Bu, kuantum kimyasasının altın standardıdır” diyor Li. Ancak CCSD(T) hesaplamalarının bilgisayar üzerinde gerçekleştirilmesi oldukça yavaş. “Elektron sayısını iki katına çıkardığınızda hesaplamalar 100 kat daha maliyetli hale geliyor.” Bu nedenle, CCSD(T) hesaplamaları genellikle 10 adet atomdan oluşan küçük moleküllerle sınırlı kalıyordu.
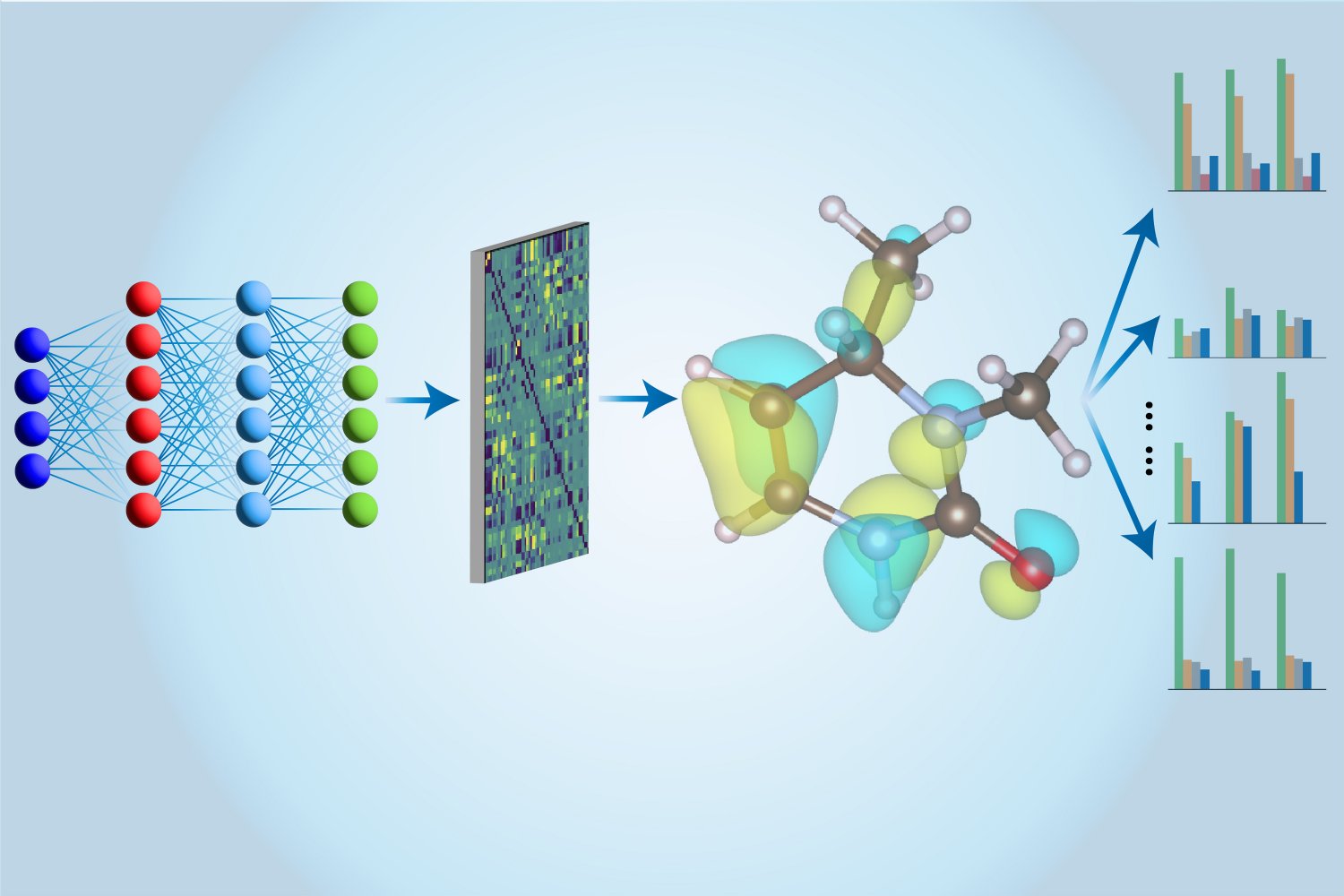
İşte burada makine öğrenimi devreye giriyor. İlk olarak, CCSD(T) hesaplamaları geleneksel bilgisayarlarda yapılıyor ve elde edilen sonuçlar Li ve ekibi tarafından özel olarak tasarlanmış bir sinir ağı ile eğitiliyor. Eğitimden sonra, sinir ağı bu hesaplamaları çok daha hızlı gerçekleştirebiliyor. Ek olarak, sinir ağı modeli, bir molekülün enerjisinin ötesinde daha fazla bilgi çıkarabiliyor. MIT doktorası öğrencisi Hao Tang, “Önceki çalışmalarda, farklı modeller kullanılarak birçok özelliği değerlendirmek için birden fazla model kullanıldı. Biz burada tek bir modelle tüm bu özellikleri değerlendirdiğimiz için buna ‘çoklu görev’ yaklaşımı diyoruz” şeklinde açıklıyor.
Test Aşaması
Li ve ekibinin geliştirdiği “Çoklu Görev Elektronik Hamiltonyen Ağı” (MEHnet), bilinen hidrokarbon moleküllerinin analizinde DFT’nin karşısında üstün performans sergiledi ve deneysel sonuçlarla yakından eşleşti. Araştırmaya katılmayan Kuzey Carolina Üniversitesi’nden malzeme keşif uzmanı Qiang Zhu, elde edilen sonuçlardan etkilendi. Zhu, “Bu yöntem, küçük bir veri seti ile etkili eğitim sağlarken, mevcut modellere kıyasla çok daha yüksek doğruluk ve hesaplama verimliliği sunuyor” diyor.
MIT’li grup, ilk olarak küçük, metalik olmayan elementleri – hidrojen, karbon, azot, oksijen ve flor – inceledi. Daha sonra ise silisyum, fosfor, kükürt, klor ve hatta platin gibi daha ağır elementlere geçtiler. Küçük moleküller üzerinde eğitim aldıktan sonra, modelin daha büyük moleküller için genelleştirilmesi sağlandı. “Önceden, hesaplamalar genellikle DFT ile yüzlerce atomu ve CCSD(T) hesaplamaları ile sadece birkaç on atomu analiz etmekle sınırlıydı,” diyor Li. “Artık binlerce atomu ve belki de on binlerce atomu işleme konusunda konuşuyoruz.”
Şu anda araştırmacılar, bilinen molekülleri değerlendiriyorlar, ancak model, daha önce görülmemiş molekülleri tanımlamak ve farklı türde moleküllerden oluşan varsayımsal materyallerin özelliklerini tahmin etmek için de kullanılabiliyor. “Teorik araçlarımızı, belirli bir kriter setini karşılayan umut verici adayları belirlemek için kullanmak istiyoruz ve ardından bunları deneysel testlere öneriyoruz” diyor Tang.
Gelecekte Neler Var?
Zhu, bu yaklaşımın yüksek hacimli moleküler tarama potansiyeli taşıdığına inandığını belirtiyor. “Bu, kimyasal doğruluğun, yeni moleküller ve istenilen özelliklere sahip malzemeleri belirlemek için kritik bir öneme sahip olduğu bir görev.” Li, önümüzdeki dönemde daha büyük molekülleri, belki de on binlerce atom içeren molekülleri analiz etme yeteneğini gösterdiklerinde, “yeni polimerler veya malzemeler” icat edebileceklerini belirtiyor. Bu tür malzemeler, ilaç tasarımı veya yarı iletken cihazlar gibi alanlarda kullanılabilir.
Sonuç olarak, Li’ye göre, gelecekte kapsam genişleyecek. “Artık sadece bir alanla sınırlı değiliz,” diyor. “Amacımız en nihayetinde, DFT’den daha düşük hesaplama maliyetiyle CCSD(T) seviyesinde doğrulukla tam periyodik tabloyu kapsamak. Bu, kimya, biyoloji ve malzeme bilimi alanında birçok problemin çözümüne olanak tanıyacak. Şu aşamada, bu yelpazenin ne kadar geniş olabileceğini söylemek zor.”
Bu çalışma, Honda Araştırma Enstitüsü tarafından desteklenmiştir. Hao Tang, Mathworks Mühendislik Bursu’ndan destek almıştır. Bu çalışmadaki hesaplamalar, kısmen Matlantis yüksek hızda evrensel atomistik simülatörü, Texas İleri Hesaplama Merkezi, MIT Süper Bulutu ve Ulusal Enerji Araştırma Bilimsel Hesaplama Tesislerinde gerçekleştirilmiştir.




